Commentary
Proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) was first described by Nasr et al. in 2004 with a subsequent study composed of a larger patient cohort a few years later [1,2]. Although more commonly occurring in older adults, PGNMID has since been reported in a wide range of age groups including children [3-5]. PGNMID is categorized as a monoclonal gammopathy of renal significance (MGRS), and is additionally included in the recently expanded concept of monoclonal gammopathy of clinical significance (MGCS), which encompasses disorders of any organ system related to underlying plasma cell or B-cell clones [6]; however, most patients with PGNMID do not show evidence of a circulating monoclonal protein or clonal plasma cell proliferation, and monoclonal gammopathy has not been reported in any pediatric patients with the disease. Herein we briefly discuss the clinical and histopathologic features of PGNMID, as well as advances in our understanding of the pathogenesis and clinical course of the disease.
Clinical Presentation
Proteinuria is the most common presenting feature of PGNMID and is present in essentially all patients, with many developing overt nephrotic syndrome [1-3]. Hematuria, typically microscopic, is also found in around 70-80% of patients [2], while the degree of renal insufficiency is variable and more common in older adults (60-80%) than in children and young adults (~50%) [1-3,5]. PGNMID also occurs more frequently in older adults, with most cases occurring in patients over 50 years of age [1,2]; however, in 2018 we reported five patients under the age of twenty with PGNMID [3], and further cases have been reported since that time [4,5]. Younger patients have an overall similar clinical presentation, although hypocomplementemia appears to be more common in pediatric patients (>50%) than in those over the age of twenty (27%) [2,3,5]. Importantly, no underlying monoclonal gammopathies were identified in any of the reported cases of PGNMID in patients under twenty years of age [3-5]. This is in contrast to older patients, in which a circulating monoclonal protein matching the glomerular deposits may be identified in around one-third of cases with IgG deposits; however, there is typically no evidence of hematologic malignancy by bone marrow biopsy at presentation or during follow-up [1,2]. Interestingly, a recent report highlighted seventeen adult patients with a light chain only variant of PGNMID [7]. These patients had a similar clinical presentation with proteinuria and hematuria, but notably a monoclonal immunoglobulin was identified in around two-thirds of patients, and a detectable plasma cell clone was identified in the bone marrow of 88% of patients [7], marking an important distinction from PGNMID with IgG deposits. No cases of light chain only PGNMID have yet been identified in the pediatric population.
Pathologic Findings
By light microscopy, PGNMID most commonly presents with a membranoproliferative-pattern of injury in both adults [2] and in pediatric patients [3-5], although some patients may show a mesangial proliferative, endocapillary proliferative, or rarely, a membranous pattern of injury [1,2,8] (Figure 1). Crescents may be present in less than a third of cases and are typically focal [2,8]. Immunofluorescence staining in PGNMID characteristically shows a pattern consistent with a complete monoclonal antibody, most commonly IgG-kappa, although lambda light chain restriction may be seen in 27-40% of adult patients [1,2] and in up to three quarters of pediatric patients [5]. Additionally, biopsies with IgM or IgA staining have infrequently been reported [5]. PGNMID demonstrates granular staining in the mesangium and along capillary loops, typically with complement, and infrequently may show a membranous pattern [2,3]. Extraglomerular deposits are not a feature of PGNMID [2]. IgG3 is the most commonly reported IgG subclass in PGNMID, followed by IgG1 and rarely IgG2 [1-5] (Figure 2). No cases of PGNMID with IgG4 deposits have been reported in the literature, which may be helpful in distinguishing PGNMID from other diseases with monoclonal IgG deposits. Electron microscopy is additionally important in distinguishing PGNMID from other glomerulonephritides with monoclonal deposits. The electron dense deposits of PGNMID are granular without the powdery, punctate appearance of those found with light chain, light and heavy chain, or heavy chain deposition disease, and they lack substructure or fibrils [1-3]. Deposits are usually distributed along the subendothelial aspect of glomerular basement membranes (GBMs), within the mesangium, and less extensively along the subepithelial surface of GBMs mirroring the pattern of staining seen by immunofluorescence (Figure 3).

Figure 1. Glomerulus with a membranoproliferative-pattern of injury characterized by marked mesangial prolfieration and frequent contour duplicaiton of basement membranes (periodic acid-Schiff, original magnification x400).
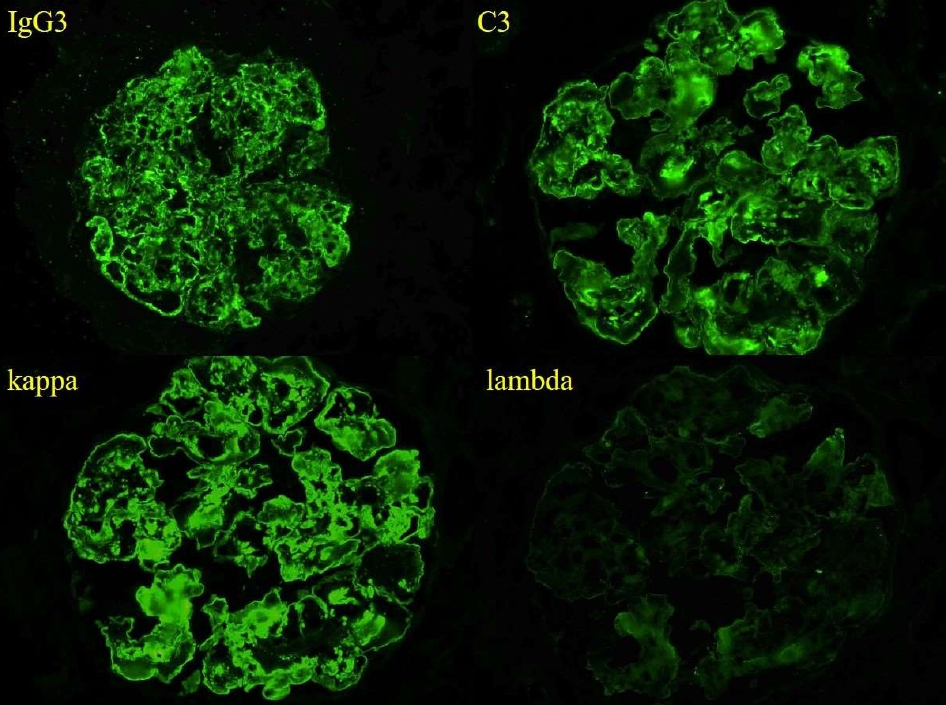
Figure 2. Immunofluroescence shows strong global granular capillary loop and mesangial staining for IgG3, C3, and kappa light chain, without significant glomerular staining for lambda light chain (IF micrographs, original magnification x400)
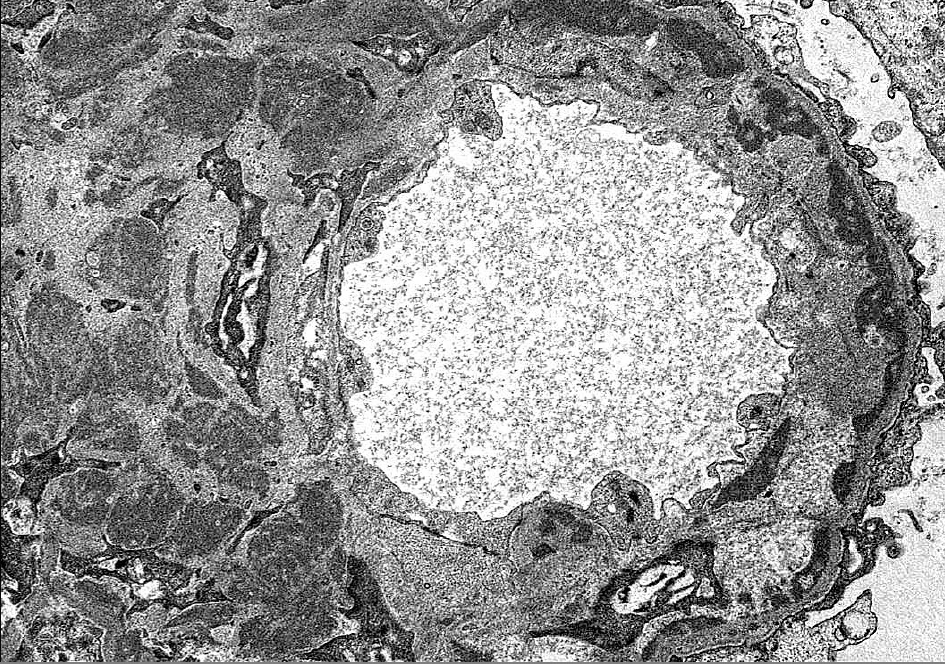
Figure 3. Granular non-Randall-type electron-dense deposits are present throughout the mesangium and along the subendothelial side of glomerular basement membranes with associated cellular interposition. There are no fibrils or substructure to deposits (Electron micrograph, original magnification x6800).
A recent series highlighted 17 cases of PGNMID with deposits composed of light chains only [7]. Like PGNMID with IgG deposits, the biopsies of patients with the light chain only variant demonstrate a membranoproliferative-pattern of glomerular injury with granular mesangial, subendothelial, and subepithelial electron-dense deposits by electron microscopy. Immunofluorescence shows staining for a single light chain, mostly kappa, with additional staining for complement C3 but no staining for IgG or immunoglobulin heavy chains.
Pathogenesis
Although PGNMID has been increasingly recognized since its initial description, the pathogenesis of the disease is still not well understood, and despite the overall unifying morphologic features, differences in the composition of deposits and the lack of a detectable circulating monoclonal protein in most patients may suggest that PGNMID represents multiple different pathologic processes. Patients with the recently described light chain only variant of PGNMID had high rates of detection of circulating monoclonal immunoglobulins with corresponding plasma cell clones on bone marrow biopsy and a good response to plasma cell-directed chemotherapy [7]; therefore, light chain only PGNMID appears to be more akin to the other dysproteinemia-related renal diseases of MGRS/MGCS such as light chain deposition disease. In contrast, patients with PGNMID with IgG deposits have much lower rates of monoclonal gammopathy and evidence of a monoclonal proliferation of plasma cells or B-cells is typically absent, thus the contribution of an underlying monoclonal gammopathy or plasma cell dyscrasia in PGNMID with IgG deposits is less clear.
The IgG deposits of PGNMID are frequently composed of IgG3, which accounts for only 8% of the total IgG in circulation and is very uncommon in plasma cell disorders [2,8]; however, IgG3 has several characteristics which allow it to easily deposit in the glomerulus and induce an inflammatory response. Compared to the other IgG subclasses, IgG3 is the most positively charged and therefore has a higher affinity for the negatively charged GBM. Additionally, IgG3 has the highest molecular weight of the IgG subclasses, restricting its ability to pass the glomerular filtration barrier. Coupled with its ability to self-aggregate through Fc-Fc interactions, this could potentially lead to aggregation of IgG3 molecules within the glomerulus. Finally, IgG3 has the greatest complement fixing capacity [2,3,5,8]. These characteristics help account for the morphologic features and complement activation seen with PGNMID, but on their own do not explain the production of monoclonal or oligoclonal immunoglobulins in adults and children without evidence of monoclonal gammopathy. While many reported adult patients do not have an obvious infectious or autoimmune trigger, it has been postulated that PGNMID arises in the setting of a clonal B-cell proliferation triggered by an immune response to an extrinsic or intrinsic antigen, and that these patients may have an underlying complement or immune system abnormality leading to persistent and excessive antibody production [2,5]. A few cases of PGNMID associated with parvovirus B19 in adults have been reported in the literature, which interestingly resolved alongside the infection [9,10]. Furthermore, an infectious trigger is suggested by Miller et al. in their report of nine pediatric patients, in which five patients had prior or active infections including influenza and group A streptococcus; however, renal disease persisted in these patients following clinical recovery from infection [5]. Ultimately, while all patients with PGNMID should be evaluated for the presence of a circulating monoclonal immunoglobulin, the pathogenesis of PGNMID in adult patients with a detectable monoclonal gammopathy likely differs from that of pediatric patients or adults without a detectable monoclonal gammopathy, and further investigation is necessary to further classify this entity.
Treatment and Clinical Outcomes
There is no established therapy for PGNMID and clinical outcomes are highly variable, reflecting the probable varying etiologies and pathogenesis of PGNMID in different patient populations. Adult patients with detectable circulating monoclonal immunoglobulins or pathogenic clones may respond to immunosuppression or directed chemotherapeutic agents including prednisone, alkylating agents, bortezomib, rituximab, and mycophenolate mofetil, among others [5,7,11], while adult and pediatric patients without a detectable monoclonal protein typically receive similar therapy with mixed results [1-5,8]. In the largest adult series, 21.9% of patients progressed to end-stage kidney disease (ESKD) in less than three years, while 37.5% had persistent renal dysfunction, 25% had partial recovery, and 12.5% experienced complete recovery [2]. In our series of five patients under the age of twenty, all but one patient who had ESKD at the time of diagnosis showed partial recovery [3], while the recent series by Miller et al. saw stable renal function in one patient with progression to ESKD in six children, four of which underwent renal transplant [5]. In those patients who undergo transplantation, PGNMID frequently recurs with monoclonal IgG deposits identical to the isotype seen in the native disease [12]. Of the four recently reported pediatric patients transplanted for PGNMID, three had documented recurrences, indicating ongoing systemic production of monoclonal immunoglobulins [5].
In conclusion, PGNMID is a relatively recently described entity which, although more common in older adults, may occur in patients of essentially any age. Categorized as an MGRS/MGCS, the disease demonstrates characteristic histopathologic features with deposition of immune complex deposits composed of monoclonal immunoglobulin molecules. Nevertheless, the pathogenesis of PGNMID is poorly understood and may in fact represent several different disease processes. Whereas a minority of adult patients with PGNMID have detectable circulating monoclonal immunoglobulins, pathogenic clones are not typically identified on bone marrow biopsy and underlying monoclonal gammopathy has not been definitively identified in any pediatric patient with PGNMID. Effective therapy for PGNMID has not yet been established, and treatment is typically aimed at an identified or suspected clonal B-cell or plasma cell population. However, most patients have long-standing renal dysfunction with nearly a quarter of adult patients progressing to ESKD, and the disease commonly recurs in allografts of both adults and children. Increased recognition of PGNMID in the pediatric setting is important, and immunofluorescence staining for kappa and lambda light chains should be routinely performed on pediatric kidney biopsies with staining for IgG subclasses utilized in all biopsies with light chain-restricted IgG deposits. Ultimately, PGNMID remains a relatively uncommon glomerular disease, and data in the pediatric setting is especially sparse. Additional studies are needed to gain a better understanding of the pathogenesis and clinicopathologic features of PGNMID in both children and adults and to establish more effective treatment options.
References
2. Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, Stokes MB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20(9):2055–64.
3. Xing G, Gillespie R, Bedri B, Quan A, Zhang P, Zhou XJ. Proliferative glomerulonephritis with monoclonal IgG deposits in children and young adults. Pediatr Nephrol. 2018;33(9):1531–8.
4. Torrealba J, Gattineni J, Hendricks AR. Proliferative glomerulonephritis with monoclonal immunoglobulin G lambda deposits: Report of the first pediatric case. Case Rep Nephrol Dial. 2018;8(1):70–5.
5. Miller P, Xiao AY, Kung VL, Sibley RK, Higgins JP, Kambham N, et al. Progression of proliferative glomerulonephritis with monoclonal IgG deposits in pediatric patients. Pediatr Nephrol. 2021;36(4):927–37.
6. Fermand J-P, Bridoux F, Dispenzieri A, Jaccard A, Kyle RA, Leung N, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. 2018;132(14):1478–85.
7. Nasr SH, Larsen CP, Sirac C, Theis JD, Domenger C, Chauvet S, et al. Light chain only variant of proliferative glomerulonephritis with monoclonal immunoglobulin deposits is associated with a high detection rate of the pathogenic plasma cell clone. Kidney Int. 2020;97(3):589–601.
8. Aucouturier P, D’Agati VD, Ronco P. A fresh perspective on monoclonal gammopathies of renal significance. Kidney Int Rep. 2021;6(8):2059–65.
9. Santana de Roberts R, Batal I, Aljareh A, Jim B. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits associated with parvovirus B19. BMJ Case Rep. 2021;14(6): e243061.
10. Fujita E, Shimizu A, Kaneko T, Masuda Y, Ishihara C, Mii A, et al. Proliferative glomerulonephritis with monoclonal immunoglobulin G3κ deposits in association with parvovirus B19 infection. Hum Pathol. 2012;43(12):2326–33.
11. Kousios A, Duncan N, Tam FWK, Chaidos A, Cook HT, Roufosse C, et al. Proliferative glomerulonephritis with monoclonal Ig deposits (PGNMID): diagnostic and treatment challenges for the nephrologist! Kidney Int. 2019;95(2):467–8.
12. Nasr SH, Sethi S, Cornell LD, Fidler ME, Boelkins M, Fervenza FC, et al. Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol. 2011;6(1):122–32.